
Materials i mètodes
Per tal de fer l'anotació de les selenoproteïnes del genoma de Pteropus alecto l'hem aliniat amb les selenoproteïnes que esperem trobar deduïdes mitjançant l'estudi evolutiu del nostre genoma. El procés l'hem automatitzat mitjançant diversos programes en llenguatge bash (clicar aquí per veure). A continuació s'exposen els procediments realitzats:
OBTENCIÓ DEL GENOMA DEL PTEROPUS ALECTO
El genoma de Pteropus alecto ha estat proporcionat pels professors de l'assignatura (/cursos/BI/genomes/project_2014/Pteropus_alecto/genome.fa). Particularment, ja es troba optimitzat per fer-ne el BLAST. També es troba disponible a la base de dades del NCBI però en aquest cas no es troba indexat.
OBTENCIÓ DE LES QUERYS
Les querys les hem obtingut a partir e la base de dades SelenoDB (www.selenodb.org), que ens proporciona en la majoria de casos diversos transcrits. Hem considerat tots els transcrits de les diverses subfamílies, tant les selenoproteïnes com les proteïnes de la maquinària que són necessàries per dur a terme la seva síntesi. D'aquesta manera ens assegurem que estem agafant totes les possibles regions d'homologia.
Les querys amb les quals hem treballat són les de l'humà (Homo sapiens), ja que és l'organisme més ben caracteritzat pel que fa a l'anotació de selenoproteïnes i de Megabat (Pteropus vampirus), l'espècie més pròxima evolutivament a Pteropus alecto indexada en el SelenoDB.
BLAST
Per començar, hem fet un BLAST (Basic Local Alignment Search Tool), un algorisme que ens permet comparar les seqüències de les selenoproteïnes d'humans o megabats (querys) amb una base de dades (el genoma de Pteropus alecto) mitjançant aliniaments locals. El tipus de BLAST utilitzat és el tBLASTn (-p tblastn), ja que es vol comparar la seqüència proteica de les querys amb els nucleòtids del genoma.
$ blastall -p tblastn -i fitxerquery.fa -d genome-fa -o query.tblastn -m8
Finalment, per poder utilitzar la informació del BLAST en processos posteriors el configurem de manera que el resultat ens surti tabulat (-m8).

SELECCIÓ DELS HITS
El BLAST ens proporciona diversos hits, seqüències de màxima similaritat. Per tal de valorar la significança d'aquests hits ens hem fixat en el valor de l'E-value que indica el nombre esperat d'aliniaments amb un score determinat que s'obtindrien per atzar en una base de dades d'una mida determinada. Així doncs, com més petit sigui aquest valor, més significatiu serà l'aliniament obtingut.
Després de realitzar el BLAST s'obté un fitxer amb el format $query.tblastn que hem emmagatzemat a /luke/blast. De cadascun dels BLAST realitzats hem seleccionat els hits que són estadísticament significatius, amb una E-value inferior a 0.001.
OBTENCIÓ DE LES REGIONS GENÒMIQUES
A partir dels hits obtinguts en el pas anterior hem delimitat les regions genòmiques de Pteropus alecto en les que podríem trobar les selenoproteïnes. Amb això, aconseguim obtenir una seqüència delimitada amb la qual treballarem més fàcilment que no pas si ho féssim amb tot el genoma.
El genoma estudiat ja havia estat indexat en diferents regions anomenades scaffolds, amb el programa Fastaindex. A continuació, gràcies al programa Fastafetch hem extret l'scaffold on trobem cadascun dels hits obtinguts en el tBLASTn i que ens eren significatius.
$ fastafetch/directori/genome.fa genome.index id_subject > nomsortida.fa
Finalment, amb el Fastasubseq hem expandit els marges de la regió indicada en els hits seleccionats tant upstream com downstream, modificant aquests paràmetres en funció de la selenoproteïna estudiada per tal d'assegurar-nos que hem agafat una regió suficientment gran i que inclou tots els exons.
$fastasubseq./fastafetch/nomsortida.fa start total_lenght > genomic.fa
PREDICCIÓ DE GENS
Fins ara hem obtingut totes les possibles regions del genoma problema amb les que podem trobar potencialment les nostres querys. Per tal de poder fer l'anotació del nostre genoma hem utilitzat dos programes: l'Exonerate i el Genewise. Ambdós programes ens permeten obtenir aliniaments més precisos.
Cal tenir en compte que en executar aquests programes no es reconeixen les Us de les querys. Per aquest motiu s'ha dissenyat un programa Perl per canviar aquests aminoàcids per X:Canvi
-
Exonerate
En primer lloc, hem utilitzat l’Exonerate per tal de definir la seqüència exònica i intrònica del genoma problema. Concretament el que fa és alinear la seqüència de la selenoproteïna amb el fragment de DNA obtingut a partir del Fastasubseq (argument: -m p2g), generant un arxiu de sortida en format GFF (argument: --showtargetgff). Utilitzarem la opció exhaustive (-E) per tal d’assegurar-nos que inclou tots els exons.
$exonerate-m p2g --showtargetgff -q query.fa -t genomic.fa -- EE > sortida.gff
Per tal de poder extreure la seqüència del cDNA a partir del fitxer GFF obtingut cal executar el programa en perl anomenat Fastaseq.gff.pl.
$ Fastaseq.gff.pl genomic.fa cDNA.gff > cDNA.fa
Finalment, el programa Fastatranslate permet poder traduir la seqüència de cDNA obtinguda a la cadena d'aminoàcids per poder-la comparar amb la query inicial.
$ fastatranslate cDNA.fa -F 1 > seq.translate.fa
El fastatranslate genera 6 possibles resultats, que corresponen als 3 marcs de lectura per a cada cadena, la forward i la reverse. Aquest darrer programa s’ha realitzat amb l’argument –F 1, de manera que hem restringit els marcs de lectura, obtenint només la primera sortida generada automàticament.
GenewiseAquest programa comparteix el mateix objectiu que l’Exonerate, dur a terme una predicció del gen, però disposa d’algorismes diferents i genera de forma automàtica la seqüència peptídica.
$ genewise -pep -pretty -cdna -gff (-trev) fitxerquery.fa genomic.fa > sortida.genewise.gff
L’argument –trev s’utilitza per buscar en la cadena reverse. Alternativament. també es pot utilitzar la opció –both, que buscarà les dues cadenes.
A continuació es compara aquesta seqüència amb la query mitjançant el t-coffee
ALINIAMENT DE PROTEÏNES T-COFFEE
A partir de l'exonerate generat en els passos anteriors hem realitzat, en primer lloc, el t-coffee (Tree-based Consistency Objective Function For alignment Evaluation). Posteriorment, i per tal de realitzar un an&agrace;lisi paral·lel de l'alineament de proteïnes hem realitzat el t-coffee a partir de la informació generada amb el genewise. El t-coffee és una eina bioinformàtica que permet realitzar alineaments múltiples de seqüències proteiques, de DNA i de RNA, gràcies a un algorisme progressiu que permet realitzar un alineament de pars tant local com global. A més, aquest programa empra un sistema d'alineament múltiple de seqüències (AMS). Nosaltres hem alineat dues seqüències proteiques, les queries humanes o les de Pteropus vampyrus amb la seqüència gènica traduïda de Pteropus alecto de la nostra predicció generada amb l'exonerate o amb el genewise.
Per l’Exonerate i fastatranslate:
$ t_coffee seq.translate.fa query.fa
Aquí es pot veure l'automatització, que inclou des de la selecció dels hits fins l'alineament amb t-coffee a partir de la sortida de l'exonerate:

A partir del fastasubseq generat a l'automatització anterior hem fet un script per automatitzar el genewise:
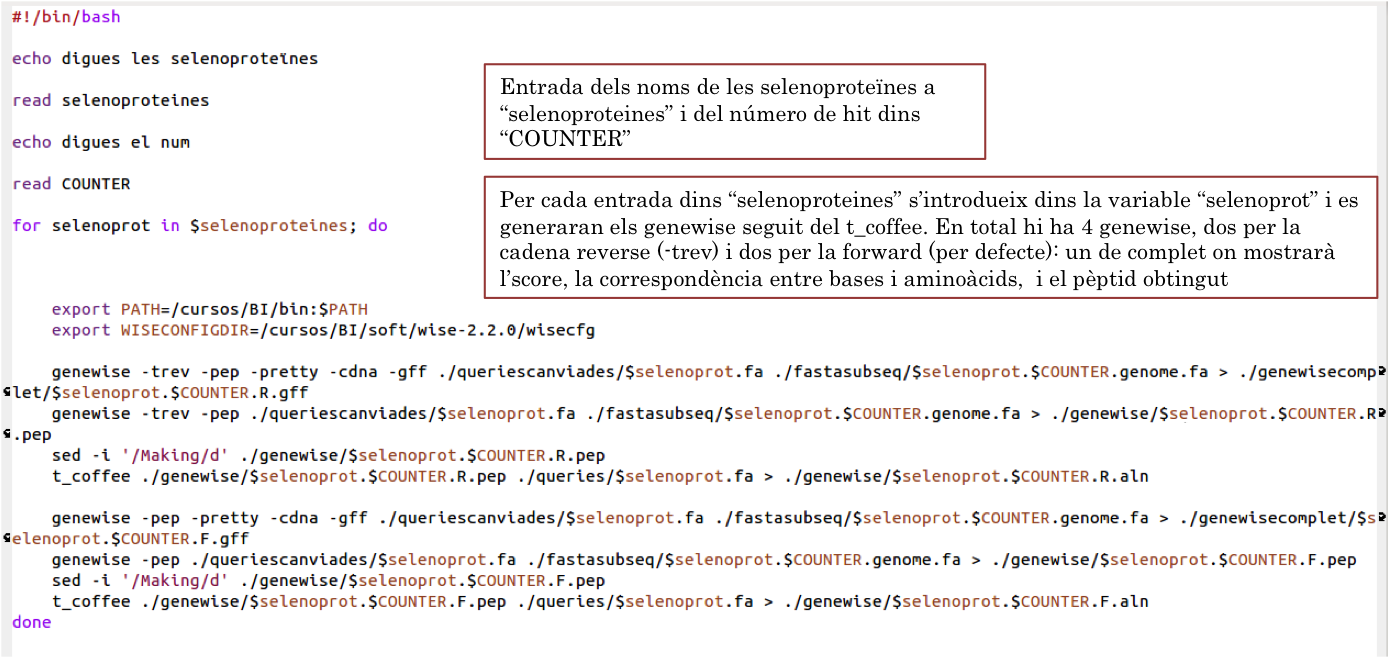
Un cop obtingut l’alineament desitjat s’ha dissenyat un programa per substituir els residus que apareixen com un asterisc per una X:IntroduirU.pl
CERCA D'ELEMENTS SECIS
A més de la cerca de les selenoproteïnes per homologia, hem realitzat la cerca d’elements SECIS (Selenocysteine Insertion Sequence) en la regió genòmica estudiada. Aquests elements són necessaris per la traducció de les selenoproteïnes i donaran suport a les prediccions realitzades. Es troben upstream del codó UGA que codifica per la selenoproteïna, a una distància que en mamífers pot variar de 200 a 5200 nucleòtids
Hem utilitzat el SECISSEarch3, que representa una millora del programa original SECISearch i es troba disponible a la pàgina web. Un cop obtingudes les prediccions, s’han analitzat els resultats valorant la posició de l’element predit, per tal de comprovar que aquest es troba a l’extrem 3’UTR de la seqüència que codifica per les selenoproteïnes predites.
SEBLASTIAN
Per tal de valorar si els anàlisis s’han realitzat correctament i poder discutir els resultats, hem utilitzat el programa Selenoprofiles, creat per Marco Mariotti del Centre de Regulació Genòmica (CRG) que està disponible a: Seblastian
Es tracta d’un nou métode que permet identificar els gens que codifiquen per selenoproteïnes. Utilitza el SECISSearch3 per predir els elements SECIS i prediu les seqüències de selenoproteïnes que estan codificades upstream d’aquests. És una eina útil tan per identificar noves selenoproteïnes com per predir-ne de noves.
ANÀLISI DELS RESULTATS
Un cop hem obtingut els alineaments del t-coffee a partir de la sortida de l’exonerate i el genewise hem dut a terme l’anàlisi dels resultats obtinguts. Per aquesta tasca, hem establert una sèrie de criteris per tal de fer l’anotació del nostre genoma. En primer lloc, hem analitzat l’alineament de proteïnes per tal d’obtenir la llargada de l'alineament (lenght), l’scaffold on es troba i la localització dins d’aquest, el nombre d’exons i introns i l’score. Concretament, hem valorat el bit score, a partir del genewise, ja que ens indica la quantitat d’informació d’un alineament sent un valor normalitzat que permetrà fer comparacions. Per contra, desestimem l’score de l’exonerate, ja que ens dóna un raw score, un valor “cru” no normalitzat que serà menys informatiu.
A partir dels alineaments també hem pogut cercar si la proteïna conté algun motiu important per la seva funció, per exemple, el motiu redox [UC]XX[UC], així com estudiar-ne els possibles esdeveniments evolutius que s’hi hagin pogut donar (duplicacions, delecions o conversions a homòlegs de cisteïna).
Si hi ha diferències entre l’alineament del genewise i l’exonerate s’intenta entendre el perquè i triar quin és el més bo.
Paral•lelament, també hem analitzat el fitxer de sortida del tBLASTn per tal d’avaluar la significança estadística dels alineaments que crèiem millors amb el valor de l'E-value, en cas que calgués discernir entre dos alineaments.
En cas de no aconseguir un bon alineament hem canviat la longitud del fastasubseq, per tal d’agafar regions més llargues o més curtes fins aconseguir el millor alineament possible.
ELABORACIÓ DE LA PÀGINA WEB
Per tal de presentar d’una manera més visual tot el treball prèviament explicat, hem realitzat una pàgina web.
Per començar s’ha fet un disseny en paper de l’estructura pensada, i s’ha plasmat en un Word amb la intenció de veure colors, mides i altres característiques de la pàgina. Un cop fet, s’ha realitzat un prototip amb html, per veure si la idea era factible o, per contra, s’havia de retocar el plantejament. El següent pas ha estat elaborar una plantilla per a les planes que seguien una mateixa estructura (clicar aquí per veure). Ja per finalitzar, s’han perfilat aquestes planes i s’han realitzat amb una estructura diferent la plana d’inici i la de contacte.
El posicionament s’ha dut a terme amb css (fulles d’estil en cascada) i taules, i els continguts amb etiquetes html i css. Per veure la fulla d’estil emprada, clicar aquí.
Per tal de veure el funcionalisme extern hem comprat un domini (grup9.net) i hem emprat el Filezilla, que és un programa d’ftp (serveix per moure fitxers). Aquesta és l’adreça: www.grup9.net/gemma.